What is PNH?
PNH is an acquired hemolytic disease caused by a genetic mutation in hematopoietic stem cells1
PNH is characterized by terminal complement–mediated intravascular hemolysis, which can lead to the devastating and potentially life-threatening consequences of thrombosis, multi-organ failure, and early mortality.2


What causes PNH?
In patients with PNH, an acquired mutation in the PIG-A gene prevents the production of GPI anchors and results in the lack, or reduced expression, of GPI-anchored complement regulatory proteins, leading to dysregulation of the complement system.1,3
Who does PNH affect?
DEMOGRAPHICS
Affects males and females equally
PNH impacts both children and adults and is believed to affect males and females in equal numbers.4,5
PREVALENCE
12 to 13 per million people
Prevalence of PNH is estimated to be 12 to 13 per million people in the general population.6
AGE OF DIAGNOSIS
Median age during the 30s

The median age at diagnosis is during the 30s.7,8
Why is diagnosing PNH so critical?
PNH is likely one of the most vicious acquired thrombophilic states known in medicine9-11
- Up to 35% of patients with PNH die within 6 years despite historical supportive care12,13,a
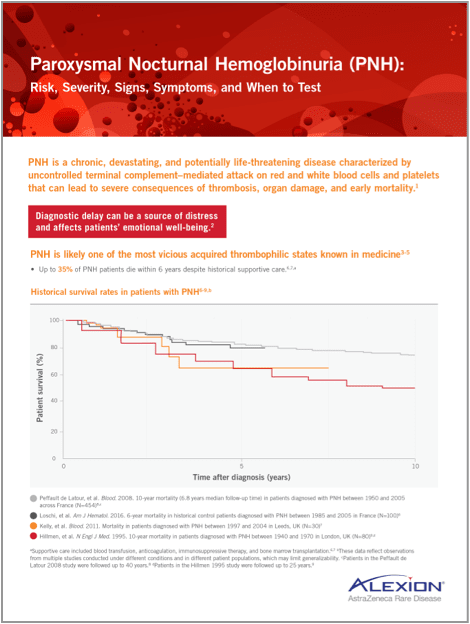
Historical survival rates in patients with PNH12-15,b
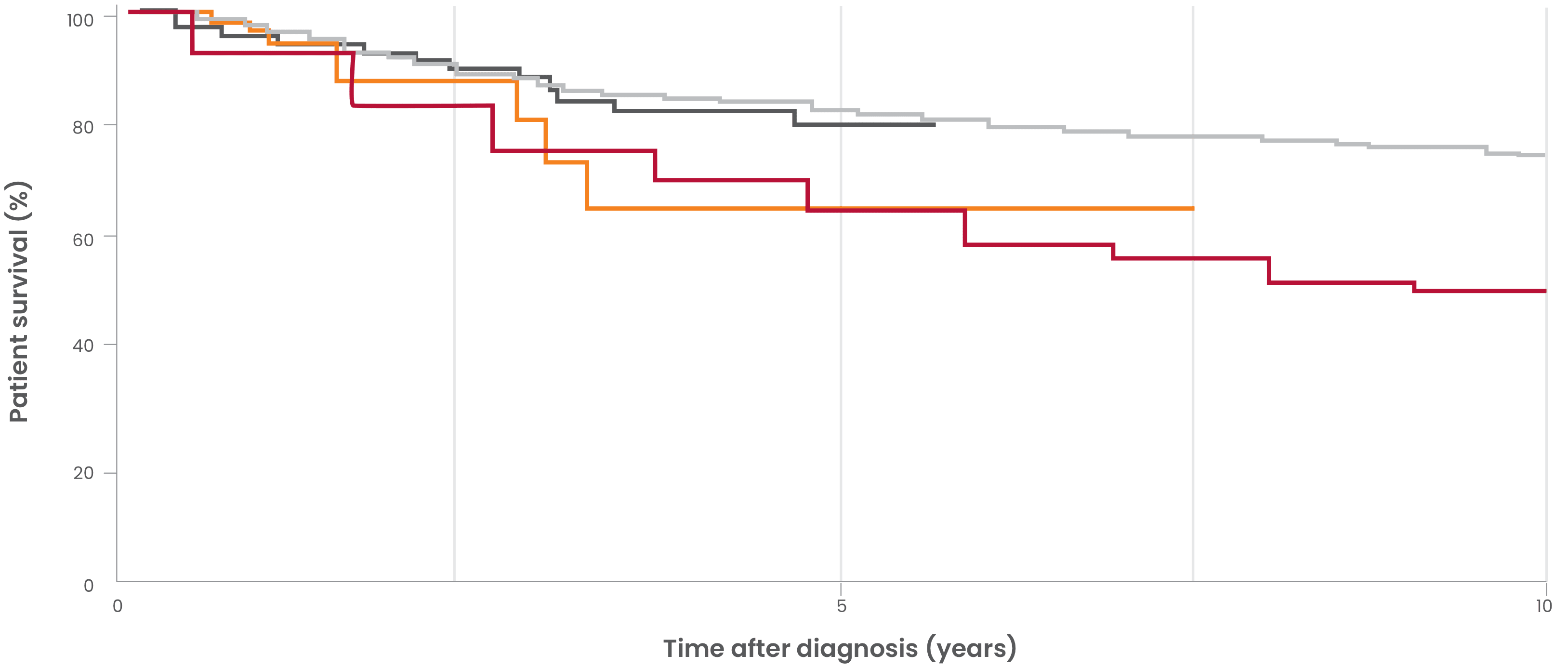
- Peffault de Latour, et al. Blood. 2008. 10-year mortality (6.8 years median follow-up time) in patients diagnosed with PNH between 1950 and 2005 across France (N=454)14,c
- Loschi, et al. Am J Hematol. 2016. 6-year mortality in historical control patients diagnosed with PNH between 1985 and 2005 in France (N=100)13
- Kelly, et al. Blood. 2011. Mortality in patients diagnosed with PNH between 1997 and 2004 in Leeds, UK (N=30)12
- Hillmen, et al. N Engl J Med. 1995. 10-year mortality in patients diagnosed with PNH between 1940 and 1970 in London, UK (N=80)15,d
- aHistorical supportive care included blood transfusion, anticoagulation, immunosuppressive therapy, and bone marrow transplantation.12,13
- bThese data reflect observations from multiple studies conducted under different conditions and in different patient populations, which may limit generalizability.
- cPatients in the Peffault de Latour 2008 study were followed up to 40 years.14
- dPatients in the Hillmen 1995 study were followed up to 25 years.15
References
- Parker CJ. Hematology Am Soc Hematol Educ Program. 2016;2016(1):208-216.
- Sharma VR. Clin Adv Hematol Oncol. 2013;11(9)(suppl 13):2-8.
- Brodsky RA. Hematology: Basic Principles and Practice. 4th ed. Churchill Livingstone; 2005:419-427.
- Bessler M, Hiken J. Hematology Am Soc Hematol Educ Program. 2008; 2008(1):104-110.
- Schrezenmeier H, et al. Ann Hematol. 2020;99(7):1505-1514.
- Jalbert JJ, et al. Blood. 2019;134(suppl 1):3407.
- Nishimura JI, et al. Medicine (Baltimore). 2004;83(3):193-207.
- NORD. Updated 2019. https://rarediseases.org/rare-diseases/paroxysmal-nocturnal-hemoglobinuria/
- Hill A, et al. Blood. 2013;121(25):4985-4996.
- Luzzatto L, et al. Br J Haematol. 2011;153(6):709-720.
- Hill A, et al. Nat Rev Dis Primers. 2017;3:17028.
- Kelly RJ, et al. Blood. 2011;117(25):6786-6792.
- Loschi M, et al. Am J Hematol. 2016;91(4):366-370.
- de Latour RP, et al. Blood. 2008;112(8):3099-3106.
- Hillmen P, et al. N Engl J Med. 1995;333(19):1253-1258.
- Brodsky RA. Blood. 2014;124(18):2804-2811.